FR-Match: Robust Matching of Cell Type Clusters from Single Cell RNA Sequencing Data
The emergence of single cell RNA sequencing (scRNAseq) is providing large amounts of single cell transcriptomics data for the unbiased quantifications of cellular heterogeneity. Though scRNAseq data have been successfully generated by many labs, less attention has been paid to how knowledge derived from these data can be integrated across studies and leveraged by the whole single cell community. In this project, we provide a user-friendly scRNAseq integration tool that uses statistical methods to map new/query cell type cluster to the reference cell types, e.g. the cell type definitions represented in semantic knowledgebase such as the Provisional Cell Ontology.
Key Features
- Input data: query and reference scRNAseq gene expression matrices with cell cluster labels
- FR-Match outputs cluster-level two-way match, one-way match, no match, and unassigned mapping results between two scRNAseq datasets
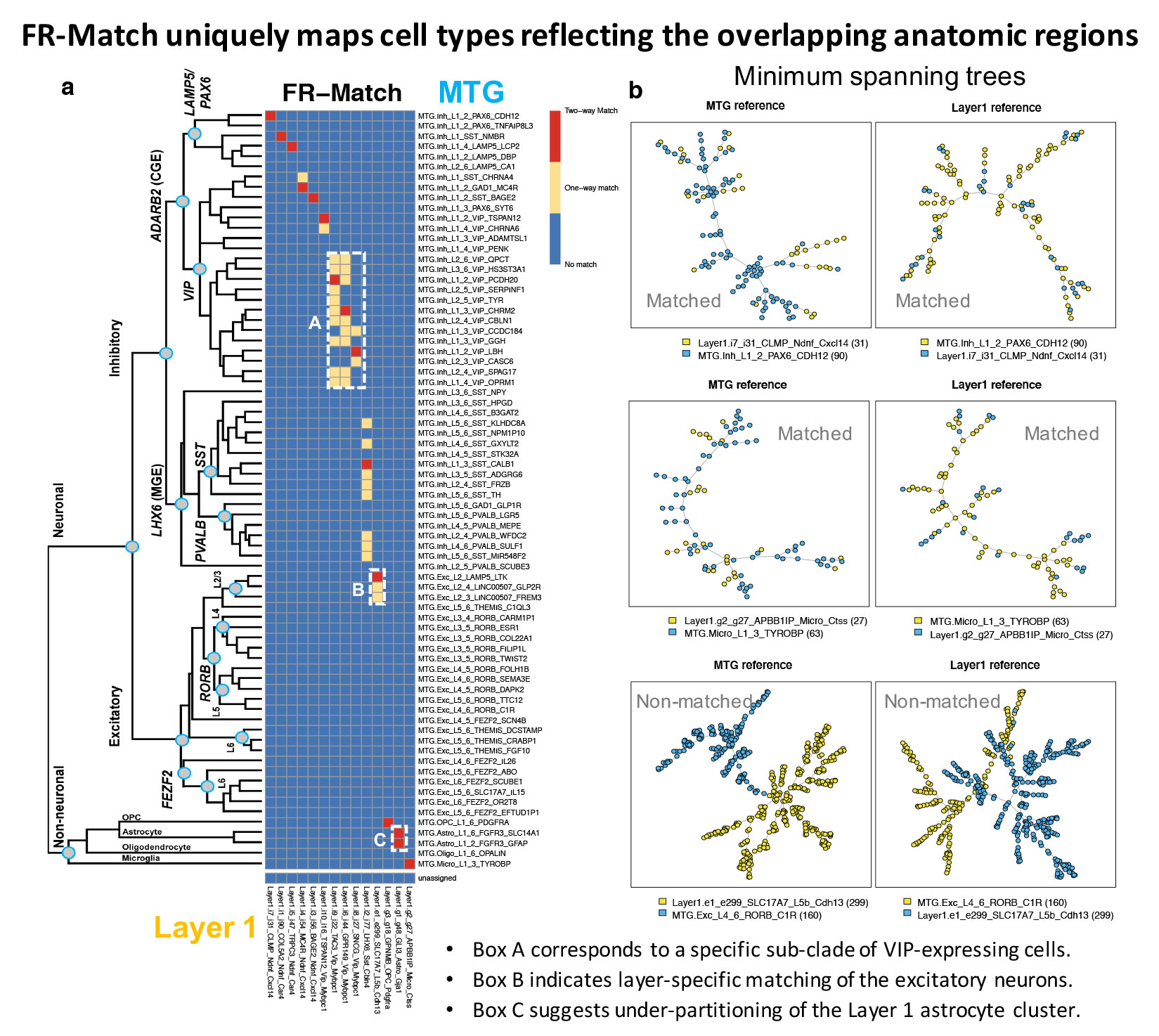
- FR-Match produces highly specific mapping of cell types between anatomically overlapping tissues (Figure a)
- FR-match provides low-dimensional visualization of matched and non-matched cell types through minimum spanning trees (Figure b)
Sample Output
Publications
Zhang et al. FR-Match: Robust Matching of Cell Type Clusters from Single Cell RNA Sequencing Data Using the Friedman-Rafsky Non-parametric Test. 2020. Briefings in Bioinformatics, accepted.
Funding
This work is funded by the JCVI Innovation Fund, the Allen Institute for Brain Science, and the Chan–Zuckerberg Initiative DAF, an advised fund of the Silicon Valley Community Foundation (2018-182730).
