High-performance comparative metagenomics
Are your carrying out large scale metagenomics analyses to identify differences among multiple sample sites? Are you looking for suitable analysis tools?
If you have not yet found the right analysis tool, you may be interested in the latest beta version of JCVI Metagenomics Reports (METAREP).
METAREP is a new open source tool developed for high-performance comparative metagenomics .
It provides a suite of web based tools to help scientists view, query, browse, and compare metagenomics annotation data derived from ORFs called on metagenomics reads or assemblies.
Users can either specify fields, or logical combinations of fields, to filter
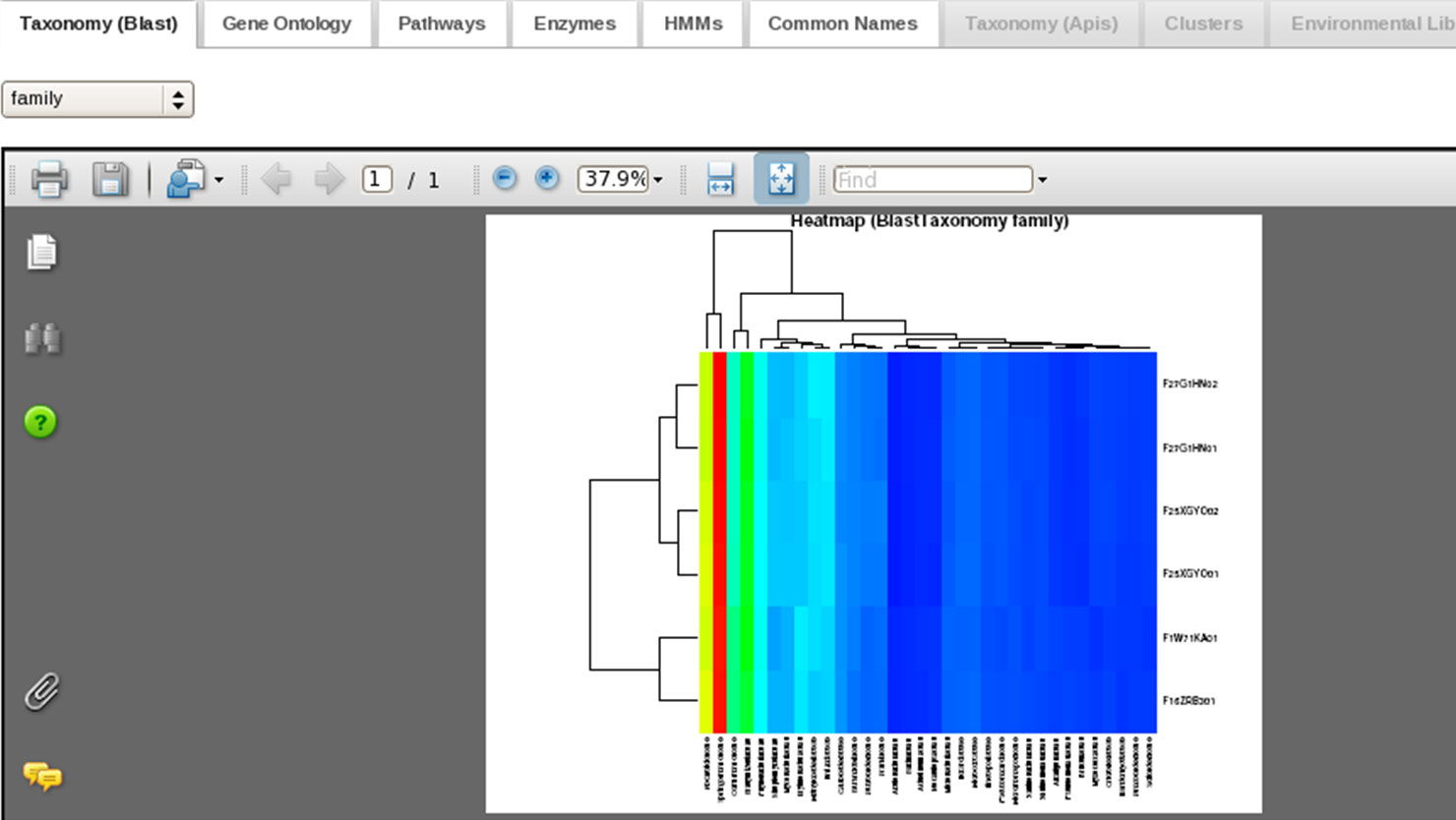
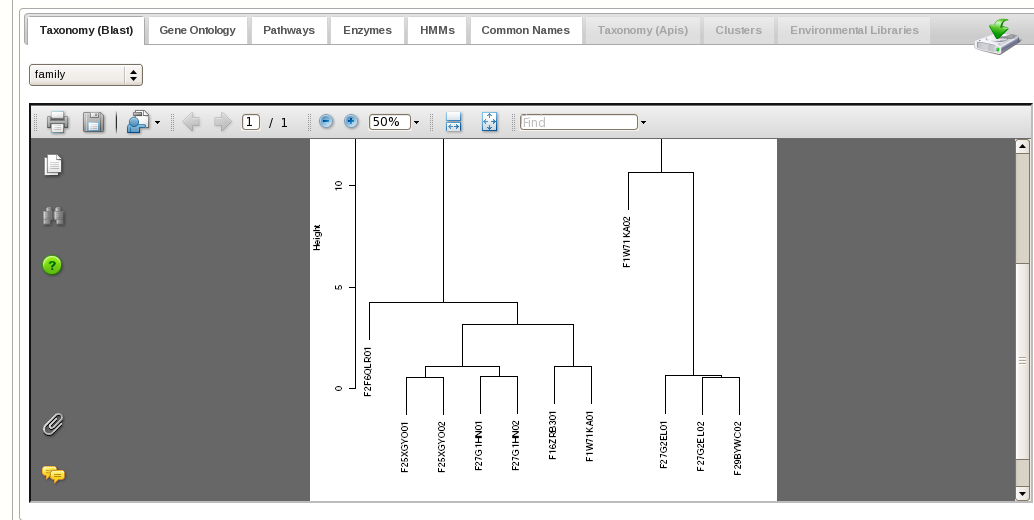
and refine datasets. Users can compare multiple datasets at various functional and taxonomic levels, applying statistical tests as well as hierarchical clustering, multidimensional scaling, and heatmaps (see image gallery).
For each of these features, tab delimited files can be exported for downstream analysis. The web site is optimized to be user friendly and fast.
Feature Summary [download Flyer]:
- Handle extremely large datasets. Uses scalable high-performance Solr/Lucene search engine (we have indexed 300 million annotation entries, but much larger volumes can be handled as shown by Hathi Trust).
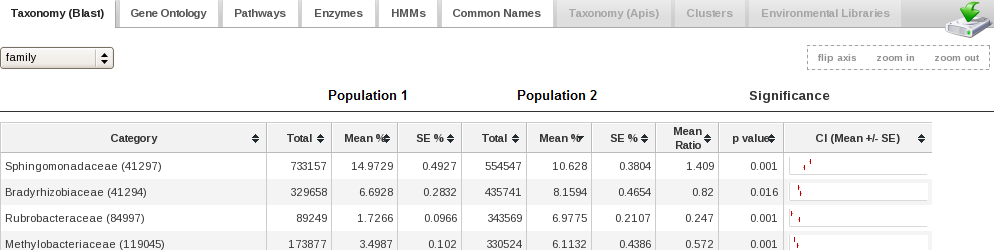
- Compare 20+ datasets at the same time. Use various compare options including statistical tests and plot options to visualize dataset difference at various taxonomic and functional levels.
- Apply statistical tests such as METASTATS (White et al.), a modified non-parametric t-test to compare two sample populations (e.g. metagenomics samples from healthy and diseased individuals).
- Export publication-ready graphics. Export heatmaps, hierarchical clustering, and multi-dimensional scaling plots in PDF format.
- Analyze KEGG metabolic pathways. Summaries include enzyme highlights on KEGG maps, pathway enzyme distributions, and statistics about pathway coverage at various pathway levels.
- Search using a SQL-like query syntax. Build your query using 14 different fields that can be combined logically.
- Drill down into data using METAREP’s NCBI Taxonomy, Gene Ontology, Enzyme Classification or KEGG Pathway browser. Install your own METAREP version.
- Flexible central configuration, METAREP and 3rd party code base is completely open source.
- Cross-link function with phylogeny. Slice your data at various taxonomic and/or functional levels. For example, search for all bacteria or exclude eukaryotes or search for a certain (GO/EC ID)/taxonomic combination.
- Generic data format. Data types that can be populated include a free text functional description, best BLAST hit information, as well as GO ID, EC ID, and HMMs.
How to analyze your own data: You can install your own METAREP version to analyze your metagenomics annotation data [download source]. We have written a comprehensive manual that describes the installation process step by step [download manual]. Since METAREP only operates on annotated data, raw sequences need to be annotated first. Supported data types that can be loaded for each sequence include functional descriptions, best BLAST hits fields (E-Value, Percent Identity, NCBI Taxon, Percent Sequence Coverage), GO, EC, and HMM assignments. The installation also contains a set of example annotations that can be imported.
Contact Us:
We would like to hear from you. If you have questions or feedback or if you wish to contribute to the METAREP open source project please send an email to metarep-support@jcvi.org
Links: